Projekt: „Zmiany w cytoszkielecie w komórkach MPS III oraz autofagia i zahamowanie syntezy glikozoaminoglikanów w leczeniu choroby Sanfilippo”, Profesor Grzegorz Węgrzyn, Uniwersytet Gdański, Polska
Raport
Opis
Cytoszkielet komórkowy jest gęstą siecią włóknistych struktur białkowych. W jego skład wchodzą trzy typy włókien: mikrotubule, filamenty aktynowe oraz filamenty pośrednie. Bierze on udział nie tylko w nadawaniu komórce kształtu i rozmiaru ale również w wielu ważnych procesach biologicznych takich jak podział mitotyczny, transport wewnątrzkomórkowy, autofagia oraz przekaźnictwo sygnału między komórkami [1]. Aby procesy te mogły być prawidłowo przeprowadzane, cytoszkielet musi wykazywać elastyczność, zgodnie z aktualnymi potrzebami komórki. Dlatego też niektóre z jego elementów – głównie mikrotubule – charakteryzują się dużą dynamiką ulegając ciągłej stabilizacji i destabilizacji poprzez polimeryzację oraz depolimeryzację tubuliny [2]. Zjawiska te występują dzięki aktywności białek towarzyszących mikrotubulom. Jednym z głównych białek należących do tej grupy i biorących udział w procesie stabilizacji mikrotubul jest białko tau. Białko to jest transferyną, której powinowactwo do mikrotubul zależy od stopnia jego fosforylacji. Nieufosforylowana forma białka wiąże się z powstającą mikrotubulą, stabilizując ją, natomiast podczas fosforylacji białko to odłącza się od mikrotubuli, co pozwala na jej destabilizację, zgodnie z chwilowym zapotrzebowaniem [3]. Czasami jednak w wyniku mutacji dochodzi do zwiększonej aktywności kinaz fosforylujących białko tau, czego skutkiem jest powstawanie jego hiperfosforylowanej formy (P-tau). Konsekwencją tego jest masowe odłączanie się tego białka od mikrotubuli i depolimeryzacja tubuliny, co prowadzi do zachwiania równowagi między stabilizacją i destabilizacją tych struktur. Odłączenie się białka tau od mikrotubul powoduje również, że do miejsc tych przyłączają się białka degradujące samą tubulinę (kataniny) [4]. Dochodzi wówczas do zaburzeń w funkcjonowaniu cytoszkieletu, a tym samym zmian w transporcie wewnątrzkomórkowym, procesie autofagii i podziale komórki. Ponadto odłączone od mikrotubuli P-tau wykazuje skłonności do tworzenia splotów spiralnie skręconych włókienek, które uszkadzają podstawowe procesy zachodzące w komórce doprowadzając do jej śmierci.
Choroby spowodowane zaburzeniami w strukturze białka tau nazywamy tauopatiami. Należą do nich między innymi choroba Alzheimera, otępienie czołowo-skroniowe i postępujące porażenie nadjądrowe [5]. Patologiczne zmiany spowodowane pojawieniem się P-tau w komórce przedstawione zostały na Ryc. 1.
Ryc. 1. Zmiany spowodowane pojawieniem się nadmiernej ilości P-tau w komórce (na podstawie [15]).
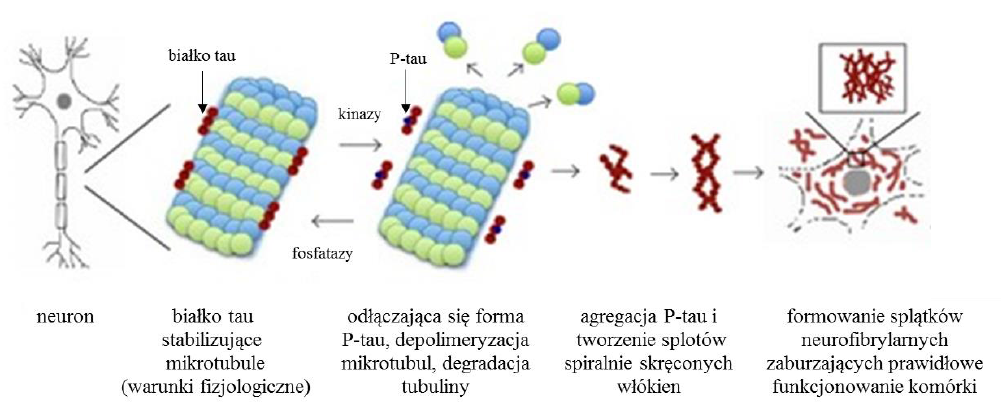
Ciekawym faktem jest, że do tej pory złogi P-tau uznano za jedną z pierwotnych przyczyn choroby Alzheimera (obok złogów β-amyloidu) mimo, że w innych chorobach neurodegeneracyjnych związanych z agregacją białek również wykazano wysoki poziom hiperfosforylowanej formy tego białka co pozwoliło na zakwalifikowanie ich jako tauopatii (np. choroba Huntingtona (HD), mukopolisacharydoza typu III (MPS III, choroba Sanfilippo) [6, 7]. Jednak o ile w przypadku choroby Alzheimera, wpływ P-tau na funkcjonowanie cytoszkieletu jest dość dobrze poznany [5], o tyle w przypadku MPS III konsekwencje dla cytoszkieletu spowodowane pojawieniem się P-tau są bardzo słabo zbadane. Niemniej jednak w literaturze pojawiają się wzmianki dotyczące obniżonego poziomu białek związanych z cytoszkieletem (badania transkryptomiczne) w przypadku MPS [8]. Znalezienie leku, który mógłby usuwać nie tylko spichrzane GAG w MPS III ale również inne tworzące się złogi, których przyczyny odkładania się w komórkach nie są jeszcze poznane (P-tau), a mają ogromne konsekwencje dla homeostazy, jest niezwykle ważne. Jest to tym bardziej istotne, że MPS zaliczane są do grupy chorób rzadkich, a wspólny cel terapeutyczny wspomógłby leczenie w całej tej grupie schorzeń, a nie tylko w pojedynczych chorobach. Kumulacja hiperfosforylowanej formy białka tau (P-tau) oraz skutkujące tym patologiczne zmiany w cytoszkielecie mogą w znacznym stopniu przyczyniać się do patogenezy choroby Sanfilippo. Możliwe, że w organizmie, w którym choroba osiągnęła zaawansowane stadium, nawet efektywne usunięcie złogów glikozoaminoglikanów (GAG) i nie dopuszczenie do ich dalszej akumulacji, nie będzie w stanie odwrócić zmian spowodowanych przez wcześniejszą akumulację P-tau i zmiany w cytoszkielecie. Zatem kluczowe dla efektywnej terapii może okazać się połączenie metody zmierzającej do zlikwidowania złogów GAG oraz zniwelowania poziomu P-tau i zmian w cytoszkielecie. Potencjalnie takie efekty mogłoby dać zastosowanie związków, które stymulują proces autofagii, ale które działają stosunkowo skutecznie a jednocześnie nie wykazują toksyczność dla komórek i organizmów. Analiza dotychczasowej literatury wskazuje na potencjalne możliwości testowania takich związków, wstępnie na hodowlach komórkowych, a następnie – w przypadku pozytywnych wyników – na modelu zwierzęcym. Zasadne jest zatem przypuszczenie, że dopiero połączenie co najmniej dwóch metod leczenia choroby Sanfilippo, polegające na obniżeniu poziomu GAG (np. poprzez zahamowanie syntezy tych związków) oraz na zwiększeniu efektywności autofagii (w celu pozbycia się 1-szo i 2-go rzędowych złogów) może dać w rezultacie odpowiednio skuteczną metodę terapeutyczną. Związki, które można przetestować jako w różny sposób stymulujące autofagię a jednocześnie potencjalnie bezpieczne stymulatory autofagii, to:
- resweratrol (inhibitor kinazy mTORC1, negatywnego regulatora procesu autofagii)
- trehaloza (aktywator białka AMPK, biorącego udział w transdukcji sygnału prowadzącego do aktywacji autofagii)
- kurkumina (stabilizator błony lizosomalnej, powodujący większą stabilność lizosomów i autofagosomów).
Jak ten projekt pomoże pacjentom chorym na Sanfilippo?
Wykonane badania wykazały, że nie tylko spichrzane GAG w MPS III ale również inne tworzące się złogi, których przyczyny odkładania się w komórkach nie są jeszcze poznane (P-tau), i moga mieć istotne konsekwencje dla postępu choroby. Podczas realizacji projektu będzie można określić czy w związku ze spadkiem poziomu P-tau, funkcjonowanie zaburzonego w MPSIII cytoszkieletu komórkowego ulegnie poprawie pod wpływem działania badanych substancji oraz czy ograniczy to powstawanie nierozpuszczalnych struktur komórkowych, które zaburzając prawidłowe funkcjonowanie komórki, nieuchronnie prowadzą do jej śmierci, co przyczynia się do neurodegeneracji w MPSIII.
Badacz prowadzacy : Prof. Grzegorz Wegrzyn
Tytuł projektu: Zmiany w cytoszkielecie w komórkach MPS III oraz autofagia i zahamowanie syntezy glikozoaminoglikanów w leczeniu choroby Sanfilippo
Lokalizacja: Uniwersytet Gdański, Polska
Status: aktywny
Start: wrzesień 2017
Comments are closed.